Examples
Example 1: Amino acid ancestral reconstruction and visualization
This example reads amino acid sequences from this FASTA file, and a phylogeny from this Newick tree file. A WAG amino acid model, augmented to explicitly model gap (ie. '-') characters, and a global substitution rate is estimated by maximum likelihood. Under this optimized model, the distribution over ancestral amino acids is constructed for each node, and visualized in multiple ways.
using MolecularEvolution, FASTX, Phylo, Plots
#Read in seqs and tree
seqnames, seqs = read_fasta("Data/MusAA_IGHV.fasta")
tree = read_newick_tree("Data/MusAA_IGHV.tre")
#Compute AA freqs, which become the equilibrium freqs of the model, and the initial root freqs
AA_freqs = char_proportions(seqs,MolecularEvolution.gappyAAstring)
#Build the Q matrix
Q = gappy_Q_from_symmetric_rate_matrix(WAGmatrix,1.0,AA_freqs)
#Build the model
m = DiagonalizedCTMC(Q)
#Set up the memory on the tree
initial_partition = GappyAminoAcidPartition(AA_freqs,length(seqs[1]))
populate_tree!(tree,initial_partition,seqnames,seqs)
#Set up a likelihood function to find the scaling constant that best fits the branch lengths of the imported tree
#Note, calling LL will change the rate, so make sure you set it to what you want after this has been called
ll = function(rate; m = m)
m.r = rate
return log_likelihood!(tree,m)
end
opt_rate = golden_section_maximize(ll, 0.0, 10.0, identity, 1e-11);
plot(opt_rate*0.87:0.001:opt_rate*1.15,ll,size = (500,250),
xlabel = "rate",ylabel = "log likelihood", legend = :none)
Then set the model parameters to the maximum likelihood estimate, and reconstruct the ancestral states.
m.r = opt_rate
#Reconstructing the marginal distributions of amino acids at internal nodes
d = marginal_state_dict(tree,m)That's it! Everything else is for visualizing these ancestral states. We'll select a set of amino acid positions to visualize, corresponding to these two (red arrows) alignment columns:
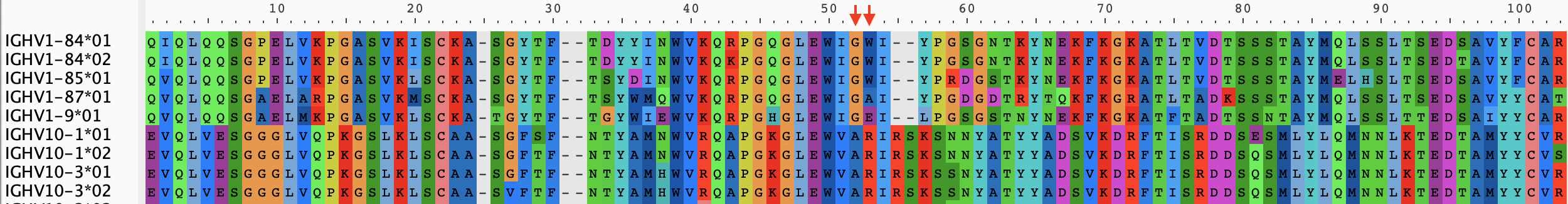
#The alignment indices we want to pay attention to in our reconstructions
motif_inds = [52,53]
#We'll compute a confidence score for the inferred marginal state
confidence(state,inds) = minimum([maximum(state[:,i]) for i in inds])
#Map motifs to numbers, so we can work with more convenient continuous color scales
all_motifs = sort(union([partition2obs(d[n][1])[motif_inds] for n in getnodelist(tree)]))
motif2num = Dict(zip(all_motifs,1:length(all_motifs)))
#Populating the node_data dictionary to help with plotting
for n in getnodelist(tree)
moti = partition2obs(d[n][1])[motif_inds]
n.node_data = Dict([
"motif"=>moti,
"motif_color"=>motif2num[moti],
"uncertainty"=>1-confidence(d[n][1].state,motif_inds)
])
end
#Transducing the MolecularEvolution FelNode tree to a Phylo.jl tree, which migrates node_data as well
phylo_tree = get_phylo_tree(tree)
node_unc = values_from_phylo_tree(phylo_tree,"uncertainty")
println("Greatest motif uncertainty: ",maximum([n.node_data["uncertainty"] for n in getnodelist(tree)]))Greatest motif uncertainty: 0.6104376723068156#Plotting, using discrete marker colors
pl = plot(phylo_tree,
showtips = true, tipfont = 6, marker_group = "motif", palette = :seaborn_bright,
markeralpha = 0.75, markerstrokewidth = 0, margins = 2Plots.cm, legend = :topleft,
linewidth = 1.5, size = (400, 800))
savefig_tweakSVG("anc_tree_with_legend.svg", pl)
pl
#Plotting, using discrete marker colors
pl = plot(phylo_tree, treetype = :fan,
showtips = true, tipfont = 6, marker_group = "motif", palette = :seaborn_bright,
markeralpha = 0.75, markerstrokewidth = 0, margins = 2Plots.cm, legend = :topleft,
linewidth = 1.5, size = (800, 800))
savefig_tweakSVG("anc_circ_tree_with_legend.svg", pl)
pl
#Plotting using continuous color scales, and using marker size to show uncertainty in reconstructions
color_scale = :rainbow
pl = plot(phylo_tree, showtips = true, tipfont = 6, marker_z = "motif_color", line_z = "motif_color",
markersize = 10 .* sqrt.(node_unc), linecolor = color_scale, markercolor = color_scale, markeralpha = 0.75,
markerstrokewidth = 0,margins = 2Plots.cm, colorbar = :none, linewidth = 2.5, size = (400, 800))
#Feeble attempt at a manual legend
motif_ys = collect(1:length(all_motifs)) .+ (length(seqs) - length(all_motifs))
scatter!(zeros(length(all_motifs)) , motif_ys , marker = 8, markeralpha = 0.75,
marker_z = 1:length(all_motifs), markercolor = color_scale, markerstrokewidth = 0.0)
for i in 1:length(all_motifs)
annotate!(0.1, motif_ys[i], all_motifs[i],7)
end
savefig_tweakSVG("anc_tree_continuous.svg", pl)
pl
Example 2: GTR+Gamma
For site-to-site "random effects" rate variation, such as under the GTR+Gamma model, we need to use a "Site-Wise Mixture" model, or SWMModel with its SWMPartition.
#Set up a function that will return a set of rates that will, when equally weighted, VERY coarsely approx a Gamma distribution
function equiprobable_gamma_grids(s,k)
grids = quantile(Gamma(s,1/s),1/2k:1/k:(1-1/2k))
grids ./ mean(grids)
end
#Read in seqs and tree, and populate the three NucleotidePartitions
seqnames, seqs = read_fasta("Data/MusNuc_IGHV.fasta")
tree = read_newick_tree("Data/MusNuc_IGHV.tre")
#Set up the Partition that will be replicated in the SWMModel
initial_partition = NucleotidePartition(length(seqs[1]))
#To be able to use unconstrained optimization, we use `ParameterHandling.jl`
initial_params = (
rates=positive(ones(6)),
gam_shape=positive(1.0),
pi=zeros(3)
)
flat_initial_params, unflatten = value_flatten(initial_params)
num_params = length(flat_initial_params)
#Setting up the Site-Wise Mixture Partition:
#Note: this constructor sets the weights of all categories to 1/rate_cats
#That is fine for our equi-probable category model, but this will need to be different for other models.
rate_cats = 5
REL_partition = MolecularEvolution.SWMPartition{NucleotidePartition}(initial_partition,rate_cats)
populate_tree!(tree,REL_partition,seqnames,seqs)
function build_model_vec(params; cats = rate_cats)
r_vals = equiprobable_gamma_grids(params.gam_shape,cats)
pi = unc2probvec(params.pi)
return MolecularEvolution.SWMModel(DiagonalizedCTMC(reversibleQ(params.rates,pi)),r_vals)
end
function objective(params::NamedTuple; tree = tree)
v = unc2probvec(params.pi)
#Root freqs need to be set over all component partitions
for p in tree.parent_message[1].parts
p.state .= v
end
return -log_likelihood!(tree,build_model_vec(params))
end
opt = Opt(:LN_BOBYQA, num_params)
min_objective!(opt, (x,y) -> (objective ∘ unflatten)(x))
lower_bounds!(opt, [-5.0 for i in 1:num_params])
upper_bounds!(opt, [5.0 for i in 1:num_params])
xtol_rel!(opt, 1e-12)
score,mini,did_it_work = NLopt.optimize(opt, flat_initial_params)
final_params = unflatten(mini)
optimized_model = build_model_vec(final_params)
LL = log_likelihood!(tree,optimized_model)
println(did_it_work)
println("Opt LL:",LL)SUCCESS
Opt LL:-3728.4761606135307Other functions also work with these kinds of random-effects site-wise mixture models:
tree_polish!(tree,optimized_model)LL: -3728.4761606135307
LL: -3728.1316616075173
LL: -3728.121005993758
LL: -3728.1202243978914
LL: -3728.1201348447107Sometimes we might want the rate values for each category to stay fixed, but optimize their weights:
#Using rate categories with fixed values
fixed_cats = [0.00001,0.33,1.0,3.0,9.0]
seqnames, seqs = read_fasta("Data/MusNuc_IGHV.fasta")
tree = read_newick_tree("Data/MusNuc_IGHV.tre")
initial_partition = NucleotidePartition(length(seqs[1]))
initial_params = (
rates=positive(ones(6)),
cat_weights=zeros(length(fixed_cats)-1), #Category weights
pi=zeros(3) #Nuc freqs
)
flat_initial_params, unflatten = value_flatten(initial_params)
num_params = length(flat_initial_params)
REL_partition = MolecularEvolution.SWMPartition{NucleotidePartition}(initial_partition,length(fixed_cats))
populate_tree!(tree,REL_partition,seqnames,seqs)
function build_model_vec(params; cats = fixed_cats)
cat_weights = unc2probvec(params.cat_weights)
pi = unc2probvec(params.pi)
m = MolecularEvolution.SWMModel(DiagonalizedCTMC(reversibleQ(params.rates,pi)),cats)
m.weights .= cat_weights
return m
end
function objective(params::NamedTuple; tree = tree)
v = unc2probvec(params.pi)
for p in tree.parent_message[1].parts
p.state .= v
end
return -log_likelihood!(tree,build_model_vec(params))
end
opt = Opt(:LN_BOBYQA, num_params)
min_objective!(opt, (x,y) -> (objective ∘ unflatten)(x))
lower_bounds!(opt, [-5.0 for i in 1:num_params])
upper_bounds!(opt, [5.0 for i in 1:num_params])
xtol_rel!(opt, 1e-12)
score,mini,did_it_work = NLopt.optimize(opt, flat_initial_params)
final_params = unflatten(mini)
optimized_model = build_model_vec(final_params)
LL = log_likelihood!(tree,optimized_model)
println(did_it_work)
println("Opt LL:",LL)SUCCESS
Opt LL:-3719.6290948420706When you have a Site-Wise Mixture (ie. REL) model, the category weights can be handled "outside" of the main likelihood calculations. This means that they can be optimized very quickly, within an objective function that is optimizing over the other parameters. The following example uses an EM approach to do this:
using Distributions, FASTX, ParameterHandling, NLopt
#Using rate categories with fixed values
fixed_cats = [(i/5)^2 for i in 1:12]
seqnames, seqs = read_fasta("Data/MusNuc_IGHV.fasta")
tree = read_newick_tree("Data/MusNuc_IGHV.tre")
initial_partition = NucleotidePartition(length(seqs[1]))
initial_params = (
rates=positive(ones(6)),
pi=zeros(3) #Nuc freqs
)
flat_initial_params, unflatten = value_flatten(initial_params)
num_params = length(flat_initial_params)
REL_partition = MolecularEvolution.SWMPartition{NucleotidePartition}(initial_partition,length(fixed_cats))
populate_tree!(tree,REL_partition,seqnames,seqs)
function build_model_vec(params; cats = fixed_cats)
pi = unc2probvec(params.pi)
m = SWMModel(DiagonalizedCTMC(reversibleQ(params.rates,pi)),cats)
return m
end
#LL for a mixture when the grid of probabilities is pre-computed
grid_ll(v,g) = sum(log.(sum((v./sum(v)) .* g,dims = 1)))
#Note: we can get away with relatively few EM iterations within the optimization cycle (in this example at least)
function opt_weights_and_LL(temp_part::SWMPartition{PType}; iters = 25) where {PType <: MolecularEvolution.MultiSitePartition}
g,scals = SWM_prob_grid(temp_part)
l = size(g)[1]
#We can optimize the category weights without re-computing felsenstein
#So it can make sense to do so within the optimization function
#Which means you don't need to optimize over as many parameters
θ = weightEM(g,ones(l)./l, iters = iters)
LL_optimizing_over_weights = grid_ll(θ,g) + sum(scals)
return θ,LL_optimizing_over_weights
end
function objective(params::NamedTuple; tree = tree)
v = unc2probvec(params.pi)
for p in tree.parent_message[1].parts
p.state .= v
end
felsenstein!(tree,build_model_vec(params))
#Optim inside optim
#We first need to handle the merge of the parent and root partitions - usually handled for us magically!
#Be careful: this example is hard-coded for a single partition
temp_part = copy_partition(tree.parent_message[1])
combine!(temp_part, tree.message[1])
θ,LL = opt_weights_and_LL(temp_part)
return -LL
end
opt = Opt(:LN_BOBYQA, num_params)
min_objective!(opt, (x,y) -> (objective ∘ unflatten)(x))
lower_bounds!(opt, [-5.0 for i in 1:num_params])
upper_bounds!(opt, [5.0 for i in 1:num_params])
xtol_rel!(opt, 1e-12)
@time score,mini,did_it_work = NLopt.optimize(opt, flat_initial_params)
final_params = unflatten(mini)
optimized_model = build_model_vec(final_params)
felsenstein!(tree,optimized_model)
temp_part = copy_partition(tree.parent_message[1])
combine!(temp_part, tree.message[1])
θ,_ = opt_weights_and_LL(temp_part, iters = 1000) #polish weights for final pass - quick
optimized_model.weights .= θ
LL = log_likelihood!(tree,optimized_model)
println(did_it_work, ":", score)
println("Opt LL:",LL)3.932150 seconds (2.38 M allocations: 2.378 GiB, 10.78% gc time, 3.28% compilation time: 7% of which was recompilation)
SUCCESS:3720.1347720900067
Opt LL:-3719.4808937732614This can be dramatically faster than trying to directly optimize over category weights when the number of categories grows. The above example took 140s with the direct approach.
Example 3: FUBAR
This example reads codon sequences from this FASTA file, and a phylogeny from this Newick tree file, and implements FUBAR.
using MolecularEvolution, FASTX, ParameterHandling, NLopt, Plots
#Read in seqs and tree
seqnames, seqs = read_fasta("Data/Flu.fasta")
tree = read_newick_tree("Data/Flu.tre")
#Count F3x4 frequencies from the seqs, and estimate codon freqs from this
f3x4 = MolecularEvolution.count_F3x4(seqs);
eq_freqs = MolecularEvolution.F3x4_eq_freqs(f3x4);
#Set up a codon partition (will default to Universal genetic code)
initial_partition = CodonPartition(Int64(length(seqs[1])/3))
initial_partition.state .= eq_freqs
populate_tree!(tree,initial_partition,seqnames,seqs)
#We'll use the empirical F3x4 freqs, fixed MG94 alpha=1, and optimize the nuc parameters and MG94 beta
#Note: the nuc rates are confounded with alpha
initial_params = (
rates=positive(ones(6)), #rates must be non-negative
beta = positive(1.0)
)
flat_initial_params, unflatten = value_flatten(initial_params) #See ParameterHandling.jl docs
num_params = length(flat_initial_params)
function build_model_vec(p; F3x4 = f3x4, alpha = 1.0)
#If you run into numerical issues with DiagonalizedCTMC, switch to GeneralCTMC instead
return DiagonalizedCTMC(MolecularEvolution.MG94_F3x4(alpha, p.beta, reversibleQ(p.rates,ones(4)), F3x4))
end
function objective(params::NamedTuple; tree = tree, eq_freqs = eq_freqs)
return -log_likelihood!(tree,build_model_vec(params))
end
opt = Opt(:LN_BOBYQA, num_params)
min_objective!(opt, (x,y) -> (objective ∘ unflatten)(x))
lower_bounds!(opt, [-5.0 for i in 1:num_params])
upper_bounds!(opt, [5.0 for i in 1:num_params])
xtol_rel!(opt, 1e-12)
@time _,mini,_ = NLopt.optimize(opt, flat_initial_params)
final_params = unflatten(mini)
nucmat = reversibleQ(final_params.rates,ones(4)) 10.596546 seconds (840.87 k allocations: 5.221 GiB, 7.45% gc time, 0.35% compilation time: 25% of which was recompilation)
4×4 Matrix{Float64}:
-9.41346 1.77048 6.85997 0.783008
1.77048 -7.24162 0.280525 5.19061
6.85997 0.280525 -8.651 1.5105
0.783008 5.19061 1.5105 -7.48412The scaling of that nuc matrix reflects the fact that the we're using a tree that was estimated under a nuc model, but here we're optimizing a codon model. No issue: the nuc rates have absorbed this scaling difference.
Now we set up a 20-by-20 grid, slicing the MG94 α and β parameters at the following values:
grid_values = 10 .^ (-1.35:0.152:1.6) .- 0.042317429393304220-element Vector{Float64}:
0.0023509298217921012
0.021069541732388508
0.047632328759699305
0.08532645148783018
0.13881657986865603
0.2147221488835822
0.3224365175323036
0.4752894025572635
0.6921964387638108
1.0
1.4367909587749033
2.05662245423022
2.9361990000358853
4.184368713262725
5.95559333316179
8.469062952630463
12.0358209216745
17.09725564569095
24.27972266134484
34.47205650419232Then we calculate the conditional likelihoods for each site. Note the 20-by-20 grid is stretched out into a length 400 vector to keep things simple. I'm avoiding reshape tricks to keep the grid structure clear.
LL_matrix = zeros(length(grid_values)^2,initial_partition.sites);
alpha_vec = zeros(length(grid_values)^2);
alpha_ind_vec = zeros(Int64,length(grid_values)^2);
beta_vec = zeros(length(grid_values)^2);
beta_ind_vec = zeros(Int64,length(grid_values)^2);
i = 1
@time for (a,alpha) in enumerate(grid_values)
for (b,beta) in enumerate(grid_values)
alpha_vec[i],beta_vec[i] = alpha, beta
alpha_ind_vec[i], beta_ind_vec[i] = a,b
m = DiagonalizedCTMC(MolecularEvolution.MG94_F3x4(alpha, beta, nucmat, f3x4))
felsenstein!(tree,m)
#This is because we need to include the eq freqs in the site LLs:
combine!(tree.message[1],tree.parent_message[1])
LL_matrix[i,:] .= MolecularEvolution.site_LLs(tree.message[1])
i += 1
end
end
prob_matrix = exp.(LL_matrix .- maximum(LL_matrix,dims = 1))
prob_matrix ./= sum(prob_matrix,dims = 1);Then we use an EM-like MAP algorithm to find the posterior grid weights, and visualize this surface:
LDAθ = weightEM(prob_matrix, ones(length(alpha_vec))./length(alpha_vec), conc = 0.4, iters = 5000);
#A function to viz the grid surface
function gridplot(alpha_ind_vec,beta_ind_vec,grid_values,θ; title = "")
scatter(alpha_ind_vec,beta_ind_vec, zcolor = θ, c = :darktest,
markersize = sqrt(length(alpha_ind_vec))/2, markershape=:square, markerstrokewidth=0.0, size=(550,500),
label = :none, xticks = (1:length(grid_values), round.(grid_values,digits = 3)), xrotation = 90,
yticks = (1:length(grid_values), round.(grid_values,digits = 3)), margin=6Plots.mm,
xlabel = "α", ylabel = "β", title = title)
plot!(1:length(grid_values),1:length(grid_values),color = "grey", style = :dash, label = :none)
end
gridplot(alpha_ind_vec,beta_ind_vec,grid_values,LDAθ)
We can see that the posterior distribution over sites is heavily concentrated at β<α. But are there any sites where β>α?
weighted_mat = prob_matrix .* LDAθ
for site in 1:size(prob_matrix)[2]
pos = sum(weighted_mat[beta_vec .> alpha_vec,site])/sum(weighted_mat[:,site])
if pos > 0.9
println("Site $(site): P(β>α)=$(round(pos,digits = 4))")
end
endSite 153: P(β>α)=0.9074
Site 158: P(β>α)=0.9266
Site 160: P(β>α)=0.9547And let's visualize one of those sites:
gridplot(alpha_ind_vec,beta_ind_vec,grid_values, weighted_mat[:,160]./sum(weighted_mat[:,160]))